Болезнь Гоше. Клинические рекомендации.
Болезнь Гоше
Оглавление
Ключевые слова
Заместительная ферментная терапия
Список сокращений
ЗФТ – заместительная ферментная терапия
АЧТВ – активированное частичное тромбопластиновое время
УЗИ – ультразвуковое исследование
МРТ – магнитно-резонансная томография
КТ – компьютерная томография
Термины и определения
?-глюкоцереброзидаза (?-глюкозидаза) — лизосомный фермент, участвующий в деградации продуктов клеточного метаболизма
Клетки Гоше – перегруженные липидами макрофаги, диаметр около 70-80 мкм, овальной или полигональной формы с бледной пенистой цитоплазмой.
Заместительная ферментная терапия (enzyme replacement therapy) – метод лечения генетических заболеваний, являющихся результатом биохимической дисфункции вследствие снижения активности фермента.
1. Краткая информация
1.1 Определение
Болезнь Гоше – наиболее частая форма из редких наследственных ферментопатий, объединенных в группу лизосомных болезней накопления.
1.2 Этиология и патогенез
Болезнь Гоше наследуется по аутосомно-рецессивному механизму. В основе заболевания лежат мутации гена глюкоцереброзидазы, локализующегося в регионе q21 на 1-й хромосоме [3, 4]. Присутствие двух мутантных аллелей гена сопровождается снижением ( 0.3;
Поиск в электронных базах данных.
Базы данных, использованных для сбора / селекции доказательств:
Доказательной базой для рекомендаций являются публикации, вошедшие в Кохрайновскую библиотеку, базы данных PUBMED и MEDLINE. Глубина поиска составляла 30 лет.
Методы, использованные для анализа доказательств:
Обзоры опубликованных мета-анализов;
Систематические обзоры с таблицами доказательств.
Методы, использованные для качества и силы доказательств:
Оценка значимости доказательств в соответствии с рейтинговой схемой доказательств (табл.П1).
Таблица П1 – Рейтинговая схема для оценки уровня достоверности доказательств
Уровни достоверности доказательств
Описание
Мета-анализы высокого качества, систематические обзоры рандомизированных контролируемых исследований (РКИ), или РКИ с очень низким риском систематических ошибок
Качественно проведенные мета-анализы, систематические обзоры или РКИ
Мета-анализы, систематические обзоры или РКИ с высоким риском систематических ошибок
Высококачественные систематические обзоры исследований случай-контроль или когортных исследований с отсутствием или очень низким риском эффектов смешивания или систематических ошибок и высокой вероятностью причинной взаимосвязи
Хорошо проведенные исследования случай-контроль или когортные исследования со средним риском эффектов смешивания или систематических ошибок и средней вероятностью причинной взаимосвязи
Исследования случай-контроль или когортные исследования с высоким риском эффектов смешивания или систематических ошибок и средней вероятностью причинной взаимосвязи
Не аналитические исследования (описания случаев, серий случаев)
Описание методики анализа доказательств и разработки рекомендаций
При отборе публикаций, как потенциальных источников доказательств, использованная в каждом исследовании методология изучалась для того, чтобы убедиться в ее соответствии принципам доказательной медицины. Результат изучения влиял на уровень доказательности, присваиваемый публикации, что в свою очередь влияет на силу, вытекающих из нее рекомендаций.
Методологическое изучение фокусировалось на особенностях дизайна исследования, которые оказывали существенное влияние на качество результатов и выводов.
С целью исключения влияния субъективных факторов каждое исследование оценивалось независимо, как минимум двумя независимыми членами авторского коллектива. Различия в оценке обсуждались на совещаниях рабочей группы авторского коллектива данных рекомендаций.
На основании анализа доказательств последовательно были разработаны разделы клинических рекомендаций с оценкой силы в соответствии с рейтинговой схемой рекомендаций (табл.П2).
Методы, использованные для формулирования рекомендаций:
Оценка значимости рекомендаций в соответствии с рейтинговой схемой (табл. П2)
Методы валидизации рекомендаций:
? внутренняя экспертная оценка;
? внешняя экспертная оценка.
Уровни доказательств.
Уровень А. Доказательства основаны на данных многих рандомизированных клинических исследований или мета-анализов, систематических обзорах.
Уровень В. Доказательства основаны на данных одного рандомизированного клинического исследования или многих нерандомизированных исследований.
Уровень С. Согласованные мнения экспертов и (или) немногочисленные исследования, ретроспективные исследования, регистры.
Уровень D. Мнение экспертов.
Самый высокий уровень рекомендаций – А.
Таблица П2 Достоверность доказательств
Систематический обзор РКИ
Систематический обзор когортных исследований
Систематический обзор «случай-контроль» исследований
Индикаторы доброкачественной клинической практики (Good Practice Points – GPPs):
Доброкачественная практика рекомендаций основывается на квалификации и клиническом опыте авторского коллектива.
Методология валидизации рекомендаций
Методы валидизации рекомендаций:
Внешняя экспертная оценка;
Внутренняя экспертная оценка.
Описание методики валидизации рекомендаций:
Настоящие рекомендации в предварительной версии были рецензированы независимыми экспертами, которых попросили прокомментировать, насколько качественно интерпретированы доказательства и разработаны рекомендации. Также была проведена экспертная оценка изложения рекомендаций и их доступности для понимания.
Предварительная версия рекомендаций представлялась и обсуждалась на заседании Экспертного Совета по проблемам болезни Гоше и научных конференциях Национального гематологического общества.
Рекомендации обсуждены и одобрены ведущими специалистами профильных Федеральных центров РФ и практическими врачами.
Окончательная редакция:
Для окончательной редакции и контроля качества рекомендации были повторно проанализированы членами авторского коллектива, которые пришли к заключению, что все существенные замечания и комментарии экспертов приняты во внимание, риск систематических ошибок при разработке сведен к минимуму.
Последние изменения и окончательная редакция данных рекомендаций были рассмотрены и утверждены 24 сентября 2014г. на заседании Мультидисциплинарного Экспертного совета по орфанным заболеваниям при Федеральных центрах МЗ РФ.
Приложение Б. Алгоритмы ведения пациента
Алгоритм диагностики болезни Гоше и ведения пациентов [20]
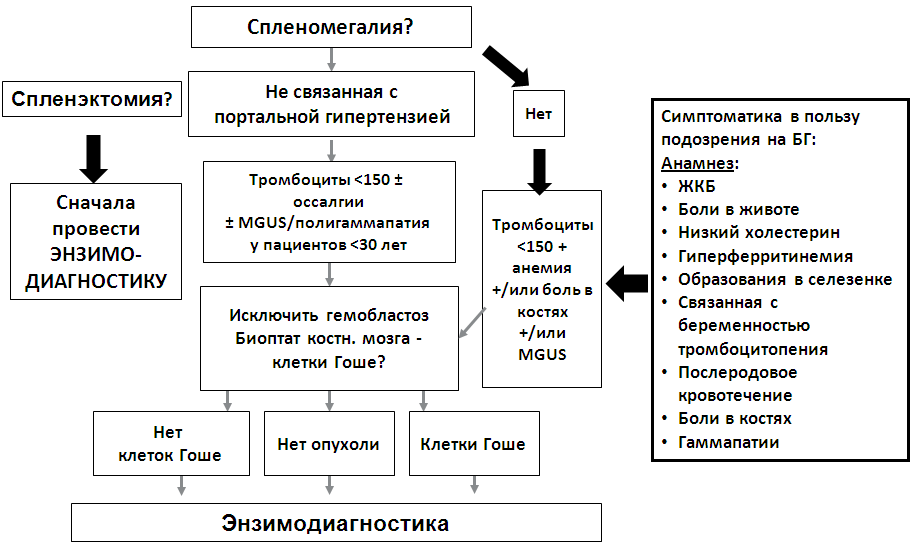
Приложение В. Информация для пациентов
Основные проявления болезни Гоше обусловлены накоплением клеток, перегруженных «шлаками», и нарушением функции этих клеток. Накопление клеток в различных органах приводит к увеличению их размеров (селезенка, печень) и/или нарушению структуры и функции (кости, костный мозг, легкие). Нарушение работы клеток (макрофагов), перегруженных шлаками, имеет следствием развитие малокровия, кровоточивости, истощения, хрупкости костей, болевых кризов. Это связано с тем, что круг «профессиональных обязанностей» макрофагов в организме человека очень широкий и включает регуляцию многих жизненно важных процессов: кроветворения, свертывания крови, обмена костной ткани и др. Наиболее типичными проявлениями болезни Гоше служат увеличение размеров селезенки и печени, развитие анемии, тромбоцитопении, хронические боли в костях или развитие внезапных приступов сильнейших болей в костях (костные кризы). Последние сопровождаюются лихорадкой и местными островоспалительными явлениями (отек, покраснение), напоминающими картину остеомиелита. Реже болезнь может впервые проявиться переломом кости вследствие незначительной травмы. Поражение костей зачастую представляет основную клиническую проблему и может привести к тяжелой инвалидизации (обездвиженность вследствие многочисленных патологических переломов, деформации костей и суставов, необходимость замены разрушенных тазобедренных или плечевых суставов).
Лечение болезни Гоше заключается в назначении заместительной ферментной терапии имиглюцеразой или велаглюцеразой – ферментами, полученными с помощью генно-инженерных технологий.
Цели лечения – предупреждение необратимого поражения костно-суставной системы и других жизненно важных органов (печень, легкие, почки); регресс или ослабление цитопенического синдрома, сокращение размеров селезенки и печени. При достижении поставленных целей назначается поддерживающее лечение в дозе 15-5 Ед/кг/инфузия (пожизненно). Заместительная ферментная терапия болезни Гоше – исключительно дорогостоящее лечение, которое во всех развитых странах мира обеспечивается специальными государственными программами. Пациенты получают лечение бесплатно. В Российской Федерации бесплатная заместительная ферментная терапия болезни Гоше стала доступной с 2007 года в рамках программы «7 нозологий».
Контакты
ФГБУ «Гематологический научный центр» МЗ РФ
Научно-клиническое отделение орфанных заболеваний
Пациентские организации:
Межрегиональная благотворительная общественная организация инвалидов «Союз пациентов и пациентских организаций по редким заболеваниям»
Рабочая группа по болезни Гоше:
Колыханов Алексей Борисович:
Межрегиональная общественная организация содействия инвалидам с детства, страдающим болезнью Гоше и их семьям:
Болезнь Гоше
Болезнь Гоше – это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью лизосомальных ферментов, накоплением гликолипидов в клеточных структурах. Симптомы определяются типом патологии. Общими признаками являются увеличение печени, селезенки, снижение свертываемости крови. При I типе выявляются нарушения со стороны костной системы: остеопороз, частые переломы, инфекции костей. При II и III типе доминирует неврологическая симптоматика: судороги, паралич, косоглазие, задержка умственного развития. Диагностика основана на биохимическом анализе дефицитарного фермента. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Общие сведения
Заболевание получило свое название по фамилии французского врача Филиппа Гоше. В 1882 году он описал симптомы и патанатомические особенности строения селезенки пациентки, которая умерла от сепсиса. Спустя несколько десятилетий при аналогичном клиническом случае Гоше определил накопление в селезенке глюкоцереброзида и недостаточность фермента глюкоцереброзидазы. Болезнь Гоше (сфинголипидоз, глюкозилцерамидный липидоз) относится к группе лизосомальных болезней накопления – наследственных патологий, при которых изменены функции клеточных органелл лизосом. Частота заболевания составляет от 1:40 тыс. до 1:70 тыс. Распространенность наиболее велика в сообществах, где допустимы браки между близкими родственниками, например, у евреев ашкенази. Носительство мутационного гена определяется примерно у 1 человека из 400.
Причины
Глюкозилцерамидный сфинголипидоз является наиболее частой формой наследственных ферментопатий. Причиной его развития считается дефект гена GBA, который кодирует фермент лизосом бета-глюкозидазу (глюкоцереброзидазу), ответственную за расщепление липидов. Наследование болезни происходит аутосомно-рецессивным способом, для формирования ферментопатии необходимо присутствие пары измененных генов: один – от матери, другой – от отца. В супружеской паре, где оба родителя – носители мутации, вероятность рождения больного ребенка составляет 25%. Риск передачи одного дефектного гена, то есть риск носительства без развития болезни в таких семьях равен 50%. При наличии в генотипе двух мутантных аллелей функция глюкоцереброзидазы снижается на 15-30% от нормального уровня.
Патогенез
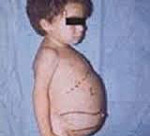
Патогенетической основой болезни является снижение каталитической активности бета-глюкозидазы. В результате нарушается процесс расщепления гликосфинголипидов (сложных соединений липидов и углеводов) до глюкозы и церамида. Аномально прогрессивное накопление макромолекул происходит в клетках, которые характеризуются повышенной скоростью их обновления – в макрофагах. Негидролизованные липиды концентрируются в лизосомах, образуются особые клетки накопления – клетки Гоше. Первичный метаболический сбой провоцирует вторичные расстройства биохимических процессов и клеточных функций. Из-за патологии жирового обмена развивается синдром активации макрофагов. Стимулируется моноцитопоэз, увеличивается содержание макрофагов в печени, селезенке, костном мозге. Это становится причиной спленомегалии, гепатомегалии, инфильтрации костного мозга. Расстройство регуляторной функции макрофагов является провоцирующим фактором цитопении, поражения костей и суставов.
Симптомы болезни Гоше
По возрасту дебюта и особенностям клинической картины выделяют три типа болезни. Первый тип наиболее распространен, имеет хронический характер течения. Симптомы чаще проявляются к 30-40 годам, реже болезнь манифестирует в детском возрасте. Увеличение размеров печени и селезенки начинается сразу после рождения, но клинически проявляется позже. Первыми признаками патологии становятся анемия, повышенная кровоточивость. Угнетение системы кроветворения сопровождается снижением уровня гемоглобина и тромбоцитов. Изменения со стороны опорно-двигательного аппарата представлены болями в костях и суставах, частыми переломами, деформациями (как правило, изменяется бедренная кость). У взрослых заметна гиперпигментация на лице и ногах: кожа темнеет, приобретает оттенок от желтоватого до желто-коричневого. Возможно появление плоских красных пятен с типичной локализацией в области вокруг глаз. Рост пациентов ниже среднего.
Второй тип болезни (острый инфантильный или острый нейропатический) встречается очень редко, развивается в промежутке от рождения до полутора лет, чаще всего симптомы дебютируют в первые три месяца жизни. Характеризуется стремительным течением, плохим откликом на лечение. На первый план выходят неврологические расстройства, спровоцированные скоплением клеток Гоше в центральной нервной системе. Дети слабо кричат, вяло сосут. Нарушен глотательный рефлекс, нередко отмечаются сбои цикла дыхания. Наблюдается заметная задержка психического и физического развития. На начальной стадии заболевания мышечный тонус снижен, через 9-12 месяцев после дебюта возникает гипертонус, особенно в мышцах шеи и конечностях. Развиваются судороги, косоглазие, спастический паралич. Печень и селезенка увеличены. Дети часто болеют тяжелой пневмонией.
Третий тип – ювенильный или подострый нейропатический. Первые признаки – увеличение селезенки и печени – возникают в 2-3 года. Полная симптоматика разворачивается в период с 6 до 15 лет. Клинические проявления поражения ЦНС включают гипертонус мышц, паралич спастического типа, косоглазие, непроизвольные спазмы, судороги, затрудненный цикл дыхания с трудностью вдоха, проблемы при глотании. Имеются расстройства психического развития: снижение интеллектуальных функций, несформированность речи и письма, эмоциональная неустойчивость, психозы. Дети отстают в половом развитии. Течение болезни неуклонно прогрессирующее.
Осложнения
Наиболее тяжелые осложнения выявляются при втором и третьем типе болезни. Поражение спинного и головного мозга приводит к нарушению дыхательного цикла, развиваются внезапные остановки дыхания, возрастает риск спазма гортани и смерти от удушья. Сниженный уровень тромбоцитов способен стать причиной обширных внутренних кровотечений. У больных с патологией первого типа распространенным осложнением является разрушение костей, их повышенная ломкость и инфекционные поражения. Ограничивается подвижность, пациенты не могут передвигаться самостоятельно, нуждаются в постороннем уходе.
Диагностика
Сбор анамнеза и физикальное обследование выполняется врачом-эндокринологом и неврологом, дополнительно назначаются консультации генетика, гематолога, офтальмолога, педиатра, психиатра. Анамнестические данные включают наличие болезни Гоше у родственников. При осмотре выявляются типичные признаки: низкий рост, патологии костей, неврологические симптомы (косоглазие, атаксия, паралич), геморрагический синдром, гиперпигментация кожи. Иногда подозрение на заболевание возникает после случайного выявления увеличенной селезенки на снимках УЗИ, угнетении кроветворной системы по данным общего анализа крови. Для подтверждения диагноза, исключения других метаболических наследственных патологий, остеомиелита, костного туберкулеза, вирусного гепатита и онкологических поражений крови проводится специфическая диагностика:
Лечение болезни Гоше
Специализированная помощь больным с первым и третьим типом болезни направлена на устранение симптомов и компенсацию первичного генетического дефекта – увеличение количества недостающего фермента, усиление катаболизма гликосфинголипидов. При 2 типе патологии терапевтические мероприятия оказываются недостаточно эффективными, усилия врачей сводятся к облегчению клинических проявлений – болей, судорог, дыхательных расстройств. Общая схема включает следующие направления:
Прогноз и профилактика
Благоприятный исход наиболее вероятен у пациентов с 1 типом заболевания – комплексный терапевтический подход позволяет нормализовать функциональность глюкоцереброзидазы, предупредить развитие осложнений, избежать инвалидизации. При 3 типе прогноз зависит от характера течения болезни, индивидуальной реакции организма на лечебные мероприятия. 2 тип имеет крайне тяжелые проявления и завершается гибелью больного. Профилактика проводится во время планирования беременности и на ее начальных сроках. Медико-генетическое консультирование рекомендуется семьям, имеющим близких родственников с данной патологией. При высоком риске передачи мутации будущему ребенку в первом триместре выполняется исследование уровня фермента в амниотической жидкости, решается вопрос о прерывании беременности.
Болезнь Гоше: орфанное заболевание в практике педиатра

Рассмотрены подходы к диагностике и лечению болезни Гоше у детей. Приведен клинический пример.
Approaches to diagnostics and treatment of Gaucher’s disease in children were considered. A clinical case is given.
В последние годы особое внимание в нашей стране уделяется проблеме ранней диагностики и лечения орфанных заболеваний.
Редкие (орфанные) заболевания — это встречающиеся с определенной частотой жизнеугрожающие или хронические прогрессирующие заболевания, приводящие без лечения к смерти или пожизненной инвалидизации пациентов. На сегодня в мире их насчитывается более 7,5 тысяч. Большинство из них обусловлено генетическими отклонениями, симптомы многих из них могут быть очевидны уже с рождения или же проявляться в детском, реже в более старшем возрасте. В 2012 г. принят Федеральный закон от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации», в котором впервые на государственном уровне введено понятие редких (орфанных) заболеваний. В нашей стране к орфанным относятся заболевания, которые имеют распространенность не более 10 случаев заболевания на 100 000 населения (статья 44). Одним из заболеваний, включенных в группу орфанных, является болезнь Гоше (код по МКБ-10 Е75.2 — «Другие сфинголипидозы»).
Болезнь Гоше (БГ) — наиболее частая форма наследственных ферментопатий из группы лизосомных болезней накопления. Тип наследования — аутосомно-рецессивный. Присутствие двух мутантных аллелей гена ассоциируется со значительным снижением (менее 30% от нормального уровня) каталитической активности β-D-глюкозидазы (глюкоцереброзидазы), что приводит к накоплению в лизосомах макрофагов глюкоцереброзида. Ген глюкоцереброзидазы картирован на хромосоме 1q21. Частота БГ в общей популяции составляет 1:40 000–1:60 000; среди евреев-ашкенази (выходцев из Восточной Европы) — 1:450–1:800 [1]. Возраст манифестации заболевания широко варьирует — от рождения до старости.
В зависимости от клинического течения выделяют 3 типа БГ:
Тип 1 является самым частым. В отличие от 2-го и 3-го типов при данном типе нервная система в патологический процесс не вовлекается [1–4].
При БГ 1-го типа основными клиническими проявлениями являются: задержка физического и полового развития, гепатоспленомегалия, геморрагический и астенический синдромы (ассоциированы с тромбоцитопенией, анемией или панцитопенией), нарушение подвижности в суставах, патологические переломы, боли в костях (костные кризы).
Наиболее ранним признаком БГ 1-го типа заболевания является спленомегалия. Селезенка может увеличиваться в размерах в 5–80 раз. По мере прогрессирования заболевания и увеличения размеров органа возможно развитие инфарктов или разрыва селезенки с возникновением фатального кровотечения. Гепатомегалия выражена в меньшей степени и развивается, как правило, в более поздние сроки заболевания, при этом функция печени не страдает. Геморрагический синдром, обусловленный тромбоцитопенией и нарушением функции тромбоцитов, проявляется в виде развития подкожных гематом, повышенной кровоточивости слизистых оболочек, длительных кровотечений после малых оперативных вмешательств, экстракции зубов, меноррагий и пр. Поражение костно-суставной системы может протекать как в виде бессимптомной остеопении и колбообразной деформации дистальных отделов бедренных костей (колбы Эрленмейера), так и в виде тяжелейшего остеопороза, сопровождающегося множественными патологическими переломами и ишемическими некрозами, приводящим к развитию вторичных остеоартрозов и тяжелых необратимых ортопедических дефектов. У трети больных отмечаются хронические боли в костях. Они могут протекать в виде костных кризов — мучительной, интенсивной боли, чаще в области нижних или верхних конечностей, сопровождающейся гиперемией и болезненностью в области суставов, снижением двигательной активности, лихорадкой, ознобом, повышением уровня маркеров воспаления (лейкоцитоз, повышенная скорость оседания эритроцитов).
При БГ 2-го типа клинические симптомы проявляются уже на первом году жизни, часто уже в первое полугодие. Заболевание имеет быстро прогрессирующее течение и характеризуется задержкой психомоторного развития с потерей ранее приобретенных навыков, нарушением глотания, часто осложняющимся аспирационной пневмонией, тризмом, билатеральным фиксированным косоглазием, гиперрефлексией, положительным симптомом Бабинского, прогрессирующей спастичностью с ретракцией шеи, приступами тонико-клонических судорог, резистентных к традиционной противосудорожной терапии, развитием гепатоспленомегалии.
При БГ 3-го типа неврологические проявления сходны с таковыми при БГ 2-го типа, но возникают, как правило, позже (в 5–6 лет и старше) и протекают менее выраженно. Для данного типа характерны окуломоторные расстройства, экстрапирамидная и мозжечковые нарушения, генерализованные тонико-клонические судороги, миоклонии. Снижение интеллекта может проявляться от незначительных изменений вплоть до тяжелой деменции. Течение заболевания медленно прогрессирующее.
Диагностика БГ основывается на выявлении характерных клинических проявлений — задержка физического и полового развития, астенический синдром, повышенная кровоточивость, боли в костях и суставах, гепатоспленомегалия, указание в анамнезе на переломы, реже — неврологическая симптоматика в виде глазодвигательной апраксии или сходящегося косоглазия, атаксии, снижение интеллекта и др., на изменениях в клиническом анализе крови — тромбоцитопения, лейкопения, анемия или панцитопения, наличие признаков гепатоспленомегалии при УЗИ органов брюшной полости, выявление диффузного остеопороза, колбообразной деформации дистальных отделов бедренных и проксимальных отделов большеберцовых костей, очагов остеолизиса, остеосклероза и остеонекроза по данным магнитно-резонансной томографии (МРТ) или рентгенографии костей скелета, патологических переломов. Проведение морфологического исследования костного мозга позволяет выявить клетки Гоше и одновременно исключить диагноз гемобластоза или лимфопролиферативного заболевания как причины цитопении и гепатоспленомегалии.
«Золотым» стандартом диагностики БГ является определение активности β-D-глюкозидазы в лейкоцитах (сухие пятна крови) — при БГ она снижена. Также диагностическим маркером может являться повышение активности хитотриозидазы — гидролитического фермента, синтезируемого активированными макрофагами в сыворотке крови, и ДНК-диагностика.
Основой патогенетического лечения БГ является проведение ферментозаместительной терапии (ФЗТ) (показана при БГ 1-го и 3-го типов). При БГ 2-го типа она не эффективна (препарат не проникает через гематоэнцефалический барьер).
На сегодняшний день на российском фармацевтическом рынке представлены следующие препараты для ФЗТ:
В связи с гетерогенностью заболевания доза ферментного препарата для каждого больного подбирается индивидуально (от 30–60 ЕД/кг на введение при 1-м типе до 120 ЕД/кг при 3-м типе) и вводится один раз в 2 недели в/в капельно. Доза может повышаться или снижаться в зависимости от степени достижения терапевтического эффекта на основании оценки клинических проявлений.
Важно помнить, что ошибочно и необоснованно при БГ проведение спленэктомии, так как это может привести к развитию тяжелых последствий: цирроза печени, деформаций костей и суставов, костных кризов, фиброза легких, проведению повторных пункций костного мозга и других инвазивных диагностических мероприятий (биопсия печени, селезенки), оперативному лечению костных кризов (часто ошибочно рассматриваются как проявления остеомиелита), назначению кортикостероидов с целью купирования цитопенического синдрома и препаратов железа (анемия при БГ носит характер «анемии воспаления»).
В качестве клинического примера приводится история развития ребенка А., 2004 г. рождения.
Диагноз: болезнь Гоше 3-го типа (E75.2) (гомозиготная мутация L444Р в гене GBA).
Осложнения: смешанный тетрапарез. Симптоматическая фокальная эпилепсия. Косоглазие содружественное сходящееся альтернирующее. Нарушение психоречевого развития. Килевидная деформация грудной клетки. Грудной кифоз III ст. S-образный левосторонний сколиоз II ст. Остеопороз. Гепатоспленомегалия. Вторичная дилатационная кардиомиопатия. НК 0 ст.
Анамнез жизни: мальчик от первой беременности, протекавшей без осложнений, роды срочные, вес при рождении 3330 г, длина 54 см. Раннее развитие без особенностей. В возрасте 2 мес выявлено снижение уровня гемоглобина до 98 г/л, по данным УЗИ — спленомегалия. Ребенок получил два курса терапии препаратами железа, однако значимого эффекта получено не было. С одного года на фоне гипертермии и фебрильных судорог стало отмечаться сходящееся косоглазие.
В возрасте 1 г 3 мес мальчик госпитализирован в гематологическое отделение, где проведена стернальная пункция. В пунктате выявлены клетки Гоше. По данным УЗИ: гепатоспленомегалия (печень +4,5 см, селезенка до +20 см из-под края реберной дуги), в анализе крови снижение уровня гемоглобина до 90 г/л; тромбоцитов — до 114 тыс. Ед/мкл. Образцы крови направлены на генетическое исследование, в результате которого установлено снижение активности фермента β-D-глюкозидазы до 1,8 нМ/мг/час и повышение уровня хитотриозидазы до 951,6 нM/мл/час. Проведена ДНК-диагностика — обнаружена гомозиготная мутация L444Р в гене GBA. Подтвержден диагноз: болезнь Гоше.
В возрасте 1 г 6 мес (04.2006 г.) ребенок начал получать ФЗТ препаратом имиглюцеразы в дозе 40 ед/кг в/в 1 раз в 14 дней. При контрольном обследовании через 6 мес от начала терапии отмечается сокращение размеров печени (до +2,5 см) и селезенки (до +12 см из-под края реберной дуги); нормализация показателей крови (гемоглобин — 121 г/л, тромбоциты — 214 тыс. Ед/мкл). Однако с 5 лет у ребенка стали отмечаться приступы повышенной сонливости с нарушением дыхания, цианозом носогубного треугольника, судорожными движениями рук, гиперсаливацией, повторяющиеся до 5–7 раз в сутки; периодичность приступов составляла 1 раз в 7–10 дней. При обследовании по данным электроэнцефалографии патологии не выявлено, на МРТ головного мозга установлена умеренно выраженная вторичная вентрикуломегалия боковых желудочков. Ребенок консультирован психоневрологом, в терапию добавлены препараты вальпроевой кислоты пролонгированного действия, леветирацетама дигидрохлорида и топирамата. Доза препарата ФЗТ повышена до 120 ед/кг.
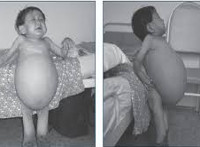
За 2 года комплексной терапии, включающей высокие дозы имиглюцеразы, у ребенка улучшились показатели физического развития (рис.), уменьшились размеры печени и селезенки, улучшилась структура их паренхимы, снизился уровень хитотриозидазы (до 527,5 нM/мл/час). Отмечается положительная динамика и со стороны неврологического статуса: исчезновение симптома Грефе, нарастание мышечного тонуса, улучшение координаторных и двигательных навыков, активная наработка словарного запаса (более 20 слов). Однако сохраняется сходящееся косоглазие.
.jpg)
Литература
Т. А. Бокова, доктор медицинских наук, профессор
ГБУЗ МО МОНИКИ им. М. Ф. Владимирского, Москва
Болезнь Гоше: орфанное заболевание в практике педиатра/ Т. А. Бокова
Для цитирования: Лечащий врач № 9/2019; Номера страниц в выпуске: 21-23
Теги: ранняя диагностика, ферментопатия, глюкоцереброзидаза